

Inadequacies of Nutrition Labeling
Rather than promoting innovation to improve health outcomes, the US government’s response to diet-associated health problems has been to focus on information and education. The idea is that if consumers are educated appropriately and have access to information about the nutritional content of foods, they will eat more healthful diets. Unfortunately, the government’s efforts have not effectively achieved their intended goals in terms of health outcomes in spite of heavy resource investment.
In 2016, the US government spent about $993.4 billion on nutrition assistance programs such as the Women and Infant Children Program and the Supplemental Nutrition Assistance Program.1 USDA, FNS “Cost of Food Distribution Programs,” 2018 https://fns-prod.azureedge.net/sites/default/files/pd/fd$sum.pdf This amount does not include all the government’s expenditures on nutrition education and outreach. Yet, despite the financial investment in healthy eating, health outcomes haven’t improved, particularly in the area of obesity.2 The Supplemental Nutrition Assistance Program, commonly known as the Food Stamp program, has been effective for low-income children in other ways—such as by decreasing anemia, failure to thrive, and nutritional deficiency. See, for example, Bong Joo Lee and Lucy MackeyBilaver, “Effects of WIC and Food Stamp Program Participation on Child Outcomes,” Children and Youth Services Review 29, no. 4 (April 2007): 501–17. For example, in 1980, 5.53 million people in the United States were diabetics (2.5 percent of the population); by 2015, that figure had jumped to 23.35 million (7.4 percent).3 Centers for Disease Control and Prevention, “Long-Term Trends in Diabetes,” Division of Diabetes Translation, April 2017. In 1990, 11.1 percent of US adults were obese; by 2016, that figure had jumped to 30.6 percent.4 “US Obesity Levels by State,” ProCon.org, updated November 8, 2018, https://obesity.procon.org/view.resource.php?resourceID=006026 Childhood obesity rates tripled from the 1970s to 2016 and quadrupled for teens over the same time period.5 Craig M. Hales et al., “Prevalence of Obesity among Adults and Youth: United States, 2015–2016” (NCHS Data Brief No. 288, National Center for Health Statistics, Hyattsville, MD, October 2017), https://www.cdc.gov/nchs/data/databriefs/db288.pdf. and “The State of Obesity,” accessed 3/15/2019, https://www.stateofobesity.org/childhood-obesity-trends/ Heart disease is also increasing.6 Kristen Fischer, “Why Heart Disease Is on the Rise in America,” Healthline, March 3, 2017, https://www.healthline.com/health-news/whyis-heart-disease-on-the-rise#1.
One problem is that educational efforts have mainly targeted highly educated and highly motivated individuals. As law professor Omri Ben-Shahar and coauthor Carl Schneider note in More Than You Wanted to Know,
- Over 40 million adults are functionally illiterate; another fifty million are only marginally literate. In one study, 40 percent of the patients could not read instructions for taking pills on an empty stomach. Innumeracy is worse. In a test of basic innumeracy only 16 percent could answer three (really) simple questions (like, how much is 1 percent of 1,000?).7Omri Ben-Shahar and Carl E. Schneider, More Than You Wanted to Know: The Failure of Mandated Disclosure (Princeton, NJ: Princeton University Press, 2014), 8.
In a 2017 article, researchers found that “many consumers have difficulty interpreting nutrition labels “and that “a sizeable proportion of the US population is deficient in health literacy.8 Persokie, Alexander et. al.” US Consumers’ Understanding of Nutrition Labels in 2013: The Importance of Health Literacy,” CDC Preventing Chronic Disease, Sept 28, 2017 A USDA study concluded that the labels had no effect on dietary intakes of total fat, saturated fat, or cholesterol (the initial dietary targets of the Nutrition Facts label).9Jayachandran N. Variyam, “Do Nutrition Labels Improve Dietary Outcomes?,” Health Economics 17, no. 6 (2008): 695–708. The study did find that label usage increased fiber and iron intake, however. Researchers have pointed out that understanding listed nutrient quantities is too complex for many consumers.10 Ilya Rahkovsky, Steve Martinez, and Fred Kuchler, New Food Choices Free of Trans Fats Better Align U.S. Diets with Health Recommendations, Economic Information Bulletin No. 95 (Washington, DC: US Department of Agriculture Economic Research Service, April 2012). Nevertheless, the government recently required the same labeling for chain restaurants, where 44 percent of calories are consumed.11 Fred Kuchler, Beyond Nutrition and Organic Labels—30 Years of Experience with Intervening in Food Labels, Economic Research Report No. 239 (Washington, DC: US Department of Agriculture Economic Research Service, November 2017).
The same concerns about usability dog the US Department of Agriculture’s MyPlate nutrition guide (a new version of the Food Guide Pyramid), which recommends what intake of food groups constitutes a healthful diet. According to researchers, most people don’t use the tool (57 percent), and it isn’t really helpful in choosing the combination of foods we should eat daily.12 F. O. Uruakpa et al., “Awareness and Use of MyPlate Guidelines in Making Food Choices,” Procedia Food Science 2 (2013): 180–86 As one nutritionist put it, “an icon isn’t going to help you eat smarter, lose weight, get in shape, or feel better about yourself,” particularly if you don’t read it, understand it, or know how to use it.13 Tina Ruggiero, quoted in “What Are the Flaws of MyPlate,” Everyday Health, accessed February 7, 2019, https://www.everydayhealth.com/diet-nutrition/experts-what-are-the-flaws-of-myplate.aspx. Terminology presents users with additional hurdles. The MyPlate icon requires users to understand what foods fall into food groups like “protein”—but not everyone does.
And yet, with nearly 45 million on specific diets each year and $33 billion spent overall trying to lose weight, the demand for help is clearly huge.14 “Weight Management,” Boston Medical Center, accessed February 7, 2019, https://www.bmc.org/nutrition-and-weight-management/weight-management. Given the problems already noted with diet regimes and consumer understanding, the next big push on the part of governments has been to develop summary measures that signal to consumers that a food is a “healthy” choice. Perhaps one of the earliest and easiest to use is the Swedish “Keyhole” icon.15 “The Keyhole,” Livsmedelsverket, National Food Agency, Sweden, reviewed February 24, 2015, https://www.livsmedelsverket.se/en/foodand-content/labelling/nyckelhalet. The Swedish National Food Agency states that “foods labelled with the Keyhole contain less sugars and salt, more fibre and wholegrain and healthier or less fat than food products of the same type not carrying the symbol.”16“The Keyhole,” Livsmedelsverket This less-is-more approach has delivered positive results. In 2010, nearly 50 percent of shoppers used the Keyhole.17 Norden, “The Keyhole: Healthy Choices Made Easy,” Nordic Council of Ministers, 2010, http://norden.diva-portal.org/smash/get/diva2:700822/FULLTEXT01.pdf Women who use the Keyhole have been shown to be thinner than those who do not, and both men and women who use it have reported lower fat intakes.18 I. Larsson, L. Lissner, and L. Wilhelmsen, “The ‘Green Keyhole’ Revisited: Nutritional Knowledge May Influence Food Selection,” European Journal of Clinical Nutrition 52 (1999): 776–80.
One alternative system, General Mills’s Nutrition Highlights, labels products with per-serving information on calories, sugars, sodium, fiber, calcium, and saturated fat. Obviously, this information is subject to the same complexity problem as the Nutrition Facts label: consumers must decide whether all the data taken together signal a healthful food choice. This becomes particularly difficult when consumers are trying to compare two alternative products.
Recently, the FDA has signaled an interest in creating a front-of-package symbol similar to the Keyhole: a standard icon that denotes “healthy.”19US Food and Drug Administration (website), “Public Meeting to Discuss FDA’s Nutrition Innovation Strategy,” updated October 12, 2018, https://www.fda.gov/Food/NewsEvents/WorkshopsMeetingsConferences/ucm611227.htm. Such a step may make government-mandated nutrition labels much more useful to the average consumer. But the Keyhole and icons like it can only provide general nutrition information and recommendations, they will not help people personalize their nutrition choices based on their genetic and health conditions.
Human Variability
Part of the problem with current nutrition-labeling systems is that recommended intakes of nutrients, calories, vitamins, and minerals ought to be personalized by age, gender, and a host of other factors.20Kuchler, Beyond Nutrition. Multiple sources of human variability affect both what consumers want to eat and what they need to eat. This variability extends to every part of the human biochemical makeup, including the processes that determine the effects of food on the body. Individual human bodies vary in the way they distribute nutrients; the way they break down complex nutrients into simpler ones; the way their cells absorb, transport, and store nutrients; and the way they excrete waste.21 Arno Motulsky, “The Genome: Nutrition and Human Variation,” in Frontiers in the Nutrition Sciences: Proceedings of a Symposium (Washington, DC: National Academy Press, 1989). Bodies that use or process nutrients differently will require different nutritional inputs.
Nutritional needs and eating practices likewise vary by a person’s culture, weight, current health condition, genetics, exercise habits, and medical history. They vary depending on whether a person takes medications, smokes, or uses dietary supplements. Finally, people’s preferences for price, convenience, and venue (e.g., eating at home versus eating out) affect their nutritional choices, as do other factors, such as where they live, how much they sleep, and how much stress they are under.
Much human variability can be measured using biomarkers. Biomarkers are metrics that can indicate the presence or risk of a disease, whether to treat the disease, and what treatment to use or even what dose to prescribe.22“Types of Biomarkers,” Personalized Medicine Coalition, Education Initiative, accessed February 8, 2019, http://www. personalizedmedicinecoalition.org/Education/Types_of_Biomarkers In medicine and clinical research, the use of biomarkers has “become so commonplace that their presence as primary endpoints in clinical trials is now accepted almost without question.”23 Kyle Strimbu and Jorge A. Tavel, “What Are Biomarkers,” Current Opinion in HIV and AIDS 5, no. 6 (2010): 463–66. For example, some biomarkers, such as a prostate-specific antigen (widely known as PSA) are used to detect diseases, in this case, prostate cancer.24Ewelina Nalejska, Ewa Mączyńska, and Marzena Anna Lewandowska, “Prognostic and Predictive Biomarkers: Tools in Personalized Oncology,” Molecular Diagnosis and Therapy 18, no. 3 (2014): 273–84. As researchers gain a more fundamental understanding of the human genome and physiological responses, they will be able to use biomarkers accurately to identify and predict more and more conditions.
At present, researchers don’t know which of the numerous input variables (e.g., specific genes, health conditions, metabolic perturbations) will prove important to prescribing diets optimized for an individual’s weight, overall health, and day-to-day functioning. However, biomarkers are already helping researchers to better understand and predict the diverse needs of different individuals. As researchers gather more data on the results of different diets, personalized nutrition, and personalized healthcare, medical devices that can incorporate all of this information and deliver it to individual consumers will become more valuable.
There have been preliminary successes. In one study, individualized diets were found “to successfully modify elevated postprandial blood glucose (a major risk factor for prediabetes and type II diabetes) and its metabolic consequences.”25 David Zeevi et al., “Personalized Nutrition by Prediction of Glycemic Responses,” Cell 163, no. 5 (2015): 1079–94 A European study showed that participants who followed Food4Me, a project gathering an international group of experts who provide personalized nutrition advice, had a higher Healthy Eating Index score than those who didn’t.26Carlos Celis-Morales et al., “Effect of Personalized Nutrition on Health-Related Behaviour Change: Evidence from the Food4Me European Randomized Controlled Trial,” International Journal of Epidemiology 46, no. 2 (2016): 578–88.
Such early successes demonstrate biomarkers’ great potential to improve health outcomes. To realize this potential, it will be necessary for researchers to create biomarkers that can be individualized and validate them. How quickly and effectively that happens will depend on how medical devices are regulated.
Confusion about Categories
The FDA currently regulates medical products differently according to what category they belong to. Products are categorized on the basis of risk, intended function, and uniqueness. Although the FDA has produced thousands of prescriptive regulations, guidance documents, and draft guidance documents describing where a product is likely to fit, its regulatory categories remain fuzzy—in fact, manufacturers must often ask the FDA to categorize their products.27 US Food and Drug Administration (website), “Premarket Notification 510(k),” updated September 27, 2018, https://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketnotification510k/default.htm. Enforcement discretion, where the agency decides when to make exceptions to its own rules, makes the categories even fuzzier.
Medical devices include a vast array of products, from innocuous devices such as toothbrushes all the way to surgically implanted devices such as pacemakers. All these products must be registered with the FDA, but the agency’s requirements beyond registration vary. Beyond registration, some require notifications that the FDA may use to prevent the product from being marketed until FDA is satisfied with the product. Still others must undergo extensive premarket clearance processes accompanied by lengthy clinical trials. There are multiple exemptions to the premarket clearance requirement, including exemptions for devices that are used by small populations or are considered to be humanitarian. (See the appendix for more information on the ways different categories of devices are regulated.) FDA has classifications established for 1,700 different generic types of medical devices with exemptions and limitations on exemptions dependent on intended use and indications for use.28FDA, “Classify Your Medical Device,” https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device
One particularly confusing issue is whether a medical device is considered (for the sake of regulation) to be a drug, a biologic (a substance made from a living organism like vaccines), or some combination of the two (for example, a prefilled syringe). As noted above, manufacturers are often unable to categorize their own products—in fact, the FDA has created an office to handle the cases in which a product could fit into multiple categories. When a manufacturer is unsure how to categorize a product, it can request a “determination” about the correct classification. The FDA will decide whether the product is a drug, a device, a biologic, or a combination product. The result is that manufacturers face uncertainty about what level of clearance they need and what type of product they are seeking to sell. The problem with this system is that boxing in innovation stifles innovation.
Medical device manufacturers face problems similar to those of companies seeking to comply with government procurement policies: “Because they’re boxed in by templates of extremely specific and confining legal and procurement criteria, vendors aren’t actually able to respond with innovative concepts and solutions, and collaborative partnerships with vendors are constricted by procurement policies.”29Erin Latham, “Innovation Can’t Happen in a Box: What Vendors Want You to Know about RFPs,” GovLoop, December 8, 2015, https://www.govloop.com/community/blog/innovation-cant-happen-box-vendors-want-know-rfps/. Medical device manufacturers, similarly, deal with uncertainty about the cost and time associated with getting their products to market. Some products may never even make it to market.
The FDA’s regulatory distinction between devices that deal directly with disease and those that don’t is also unhelpfully imprecise. Devices that “diagnose, cure, mitigate, treat, or prevent disease, or affect the structure or function of humans (or other animals),” are subject to rigorous regulatory rules. Devices that supposedly do none of those things are exempt from regulation (except for registration), but the line between the two categories is subjective. For example, 60 percent of US adults have at least one chronic condition that affects their physical or mental health. Examples include high blood pressure, high cholesterol, anxiety, arthritis, diabetes, and heart disease.30 Christine Buttorff, Teague Ruder, and Melissa Bauman, Multiple Chronic Conditions in the United States (Santa Monica, CA: RAND, 2017). In addition, everyone has a genetic predisposition toward some fatal illness or has a disease biomarker. As outlined previously, biomarkers include metrics like body temperature, blood sugar, and genetic variants that can serve as indicators that an individual either has a disease or is predisposed to a disease.31 Janet Woodcock, “Two Recent Scientific Advances Underscore an Encouraging Future for Precision Medicine at FDA,” FDA Voice, July 11, 2017.
Everything you eat, every aspect of your lifestyle (including exercise, sleep, and amount of stress-producing activity) will affect your predisposition for disease in some manner. This means that any time a device makes a health measurement—or provides advice about which foods to eat or how to exercise—it could qualify as a medical device that requires more stringent regulation.
The FDA’s current approach to regulating digital health technologies derives, at least in part, from the 21st Century Cures Act. That law is designed to “accelerate medical product development.”32Sullilvan,Thomas, “21st Century Cures Act: Implementation Update,” Policy and Medicine,” A Rockpointe Publication, Feb 17, 2019 https://www.policymed.com/2019/02/21st-century-cures-act-implementation-update.html Currently, medical devices that are “for maintaining or encouraging a healthy lifestyle and [are] unrelated to the diagnosis, cure, mitigation, prevention or treatment of a disease or condition” are not subject to FDA oversight. Such products are considered “general wellness devices,” according to FDA guidance documents.33FDA, “General Wellness: Policy for Low Risk Devices,” July 29, 2016. https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm429674.pdf Thus, these products are supposed to be free of the regulatory requirements imposed on FDA-regulated products.
Because everyone has adverse health conditions of some sort, everything that enters the body, including food, has the potential to cure, mitigate, treat, or prevent disease or to affect physical or mental health.34 In fact, some foods, called “medical foods,” are specially formulated to manage diseases. An example is Lofenalac, a medical food that has a full complement of nutrients except the offending nutrient. The same goes for environmental factors and exercise. This fact makes it difficult to create a meaningful distinction that is neither vague nor subjective between devices that serve as general wellness devices and those that relate to “the diagnosis, cure, mitigation, prevention or treatment of a disease or condition.”35 21st Century Cures Act of 2016, Pub. L. No. 114-255. This lack of clarity allows the FDA to use its discretion when choosing whether to subject a product to its oversight. The same discretion creates uncertainty for innovators about the stringency and burden of their regulatory requirements.
On December 8, 2017, the FDA issued draft guidance to indicate how it is going to comply with the 21st Century Cures Act by regulating general wellness devices.36 US Food and Drug Administration, “Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act: Draft Guidance for Industry and Food and Drug Administration Staff,” December 8, 2017. That guidance was intended to update past practices as promulgated in several previous documents.37 These documents included US Food and Drug Administration, “General Wellness: Policy for Low Risk Devices: Guidance for Industry and Food and Drug Administration Staff,” July 29, 2016; US Food and Drug Administration, “Mobile Medical Applications: Guidance for Industry and Food and Drug Administration Staff,” February 9, 2015; and US Food and Drug Administration, “Guidance for Industry, FDA Reviewers and Compliance on Off-the-Shelf Software Use in Medical Devices,” September 9, 1999. Agency guidance is not legally binding and can be another source of regulatory uncertainty for companies trying to determine their regulatory requirements.
General wellness devices are devices that encourage a “general state of health or healthy activity.” They are not subject to medical device regulations.38 US Food and Drug Administration, “General Wellness,” quoted in US Food and Drug Administration, “Changes to Existing Medical Software Policies.” But any such device that has software functions “related to the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition” would be covered by the medical device regulations.39US Food and Drug Administration, “Changes to Existing Medical Software Policies.” By using the term “software functions” rather than apps, the law “allows FDA more flexibility in deciding how to regulate complex products that may contain multiple software functions.”40Moe Alsumidaie, “FDA Releases New Digital Health Draft Guidance Documents,” Applied Clinical Trials, December 19, 2017. Software functions are more general than “apps” and will give FDA greater flexibility as to what it chooses to regulate.
The FDA refers to the foregoing regulatory activity as “enforcement discretion.” However, as one group put it, the new guidance does not “address dynamic digital health solutions . . . incorporating machine learning, artificial intelligence, or blockchain technologies.” Again, because FDA is vague in its guidance with respect to these new products, the discretion allowed under the 21st Century Cures Act leaves manufacturers uncertain.41Christina Kuhn et al., “FDA Outlines Updated Approach to Regulating Digital Health Technologies,” Covington Digital Health, December 20, 2017, https://www.covingtondigitalhealth.com/2017/12/fda-outlines-updated-approach-to-regulating-digital-health-technologies/. The FDA can also rescind exclusions. For example, a device is not excluded from regulation if it receives a signal from an electronic monitoring device such as a blood pressure monitor.42Kuhn et al., “FDA Outlines Updated Approach.” This inhibits many potential innovations that are based on modern health technologies.
The FDA notes that general wellness devices are not in fact excluded from the “medical device” category—they are merely exempted (at least currently) from the regulatory requirements that apply to medical devices. Examples of general wellness devices include software that makes “healthy lifestyle claims,” such as claims to help with “weight management, physical fitness, relaxation or stress management, mental acuity, self-esteem, sleep management, or sexual function.”43FDA, “Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act, Draft Guidance for Industry and Food and Drug Administration Staff,” Dec 8, 2017, p.8. Such software is not treated as a medical device when it is not involved in “the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition.”44 IBID. In other words, a device that monitors and records food consumption to manage weight or alert users to an unhealthy dietary choice is considered to be a medical device, but the FDA does not currently intend to enforce its regulatory requirements on such devices. The same goes for mobile medical apps.
This situation gives rise to considerable uncertainty about how and when products will be regulated—and when the FDA might change its mind. The outcome of such uncertainty is evident in the FDA’s historical record. The FDA’s intention to enhance consumer safety may have unintentionally caused consumer safety losses, in the form of safety-improving innovations that never reached the marketplace.45 See, for example, STAT, “Medical devices for pain, other conditions have caused more than 80,000 deaths since 2008, Nov 25, 2018. https://www.statnews.com/2018/11/25/medical-devices-pain-other-conditions-more-than-80000-deaths-since-2008/ The regulatory structure intended to allow only safe products to reach consumers also systematically disincentivizes innovation in the medical-device space.
Trend toward Greater Regulation
Although the FDA has exercised discretion in the past, the overall trend has been toward more stringent regulatory requirements rather than liberalization. Historically, the FDA has increased its control by discrete degrees rather than by steady expansion: regulatory stringency tends to increase in leaps, particularly following anecdotal reports of regulatory failure.46Robert Higgs, “Wrecking Ball: FDA Regulation of Medical Devices” (Policy Analysis No. 235, Cato Institute, August 7, 1885), https://object.cato.org/pubs/pas/pa235.pdf.
For example, the FDA owes its expanded control over medical devices to problems with the drug Sulfanilamide.47 This is explained in: Richard Williams, Marc Joffe, and Ariel Slonim, “Health Options Foreclosed: How the FDA Denies Americans the Benefits of Medical Research” (Mercatus Working Paper, Mercatus Center at George Mason University, Arlington, VA, September 2016). In the case of drugs, “the agency [has] shifted from ensuring honest and accurate drug information to providing strict regulatory drug oversight. This stricter regulatory scheme imposes significant costs, which are generally justified by claims that such a system ensures consumer safety.”Williams, Joffe, and Slonim, “Health Options Foreclosed.”
The FDA’s modus operandi has changed dramatically over its history. One author explains, “What started as a fairly simple regime of after-the-fact policing aimed at substandard foods and drugs has morphed into a complex set of product licensing requirements.”48Lars Noah, “The Little Agency That Could (Act with Indifference to Constitutional and Statutory Structures),” Cornell Law Review 93 (2008): 901–25. In light of this shift, FDA regulations should be evaluated to determine whether they are still improving health and safety outcomes.
The FDA’s growing control over the products it regulates comes from both internal FDA decisions and congressional grants of additional regulatory authority. On the basis of its history, the FDA can be expected to regulate AI-powered nutrition devices more strictly over time—unless it confronts a politically compelling reason not to do so. As these devices incorporate more useful functions, such as the ability to monitor health biomarkers, they will be perceived as more risky. The FDA will use this as a justification to regulate them more stringently, and the increased regulation will block or delay devices that would otherwise yield beneficial outcomes.
As technology advances, manufacturers of devices that now offer only nutrition advice are likely to modify their devices, incorporating features that help users address diseases or biomarkers of disease. Without these functions, health devices will not be anywhere near as useful to improve health. If devices are unable to incorporate these health-improving features because of the regulatory burden or because of regulatory uncertainty, consumers will lose out on health benefits.
Pre-certification of Digital Health Firms
The FDA is conducting a pilot program for software whereby it pre-certifies “eligible digital health developers who demonstrate a culture of quality and organizational excellence based on objective criteria.”49 US Food and Drug Administration, “Digital Health Innovation Action Plan,” Center for Devices and Radiological Health, Digital Health Program, n.d. In other words, they plan to approve organizations instead of products. This program will clearly affect competition in the medical-device marketplace. One entrepreneur explains,
- For those few companies who do make it through [pre-certification], they have an enormous competitive advantage. We won’t have to worry about the usual scenario: Two guys from Stanford in a garage inventing some app that instantly takes away all our business. We only have to worry about the big players—who might just buy us out instead of competing with us.50 Jonathan Kay, “Technology: How Do You Regulate a Self-Improving Algorithm?,” Atlantic, October 25, 2017, https://www.theatlantic.com/technology/archive/2017/10/algorithms-future-of-health-care/543825/.
The factors the FDA has said it will consider using to certify companies include “company size, demonstrated record of quality and organizational excellence, clinical focus area and the risk profile of the product.”51 US Food and Drug Administration, “FDA Selects Participants for New Digital Health Software Precertification Pilot Program,” news release, September 26, 2017, https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm577480.htm. As of June 2018, the pilot program is reviewing firms to show that they have “demonstrated capabilities to build, test, monitor, and proactively maintain and improve the safety, efficacy, performance, and security of their medical device software products, so that they meet or exceed existing FDA standards of safety and effectiveness.”52 US Food and Drug Administration, Developing Software Precertification Program: A Working Model, version 0.2, June 2018 These developments seem to validate a longstanding concern that the FDA serves large firms at the expense of smaller ones.53 Sydney Lupkin, “A Look at How the Revolving Door Spins from FDA to Industry,” NPR, September 28, 2016, https://www.npr.org/sections/health-shots/2016/09/28/495694559/a-look-at-how-the-revolving-door-spins-from-fda-to-industry
The FDA has not yet announced whether the pre-certification program will take the place of premarket product approval. As Bakul Patel, the FDA’s associate director for digital health, says, “We are evolving that space.”5954 Jonathan Kay, “Technology: How Do You Regulate a Self-Improving Algorithm?,” Atlantic, October 25, 2017, https://www.theatlantic.com/technology/archive/2017/10/algorithms-future-of-health-care/543825/. Bakul goes on to say that agency officials will focus their inspections on “the software developer or digital-health technology developer, rather than primarily at the product.”55 Jonathan Kay, “Technology: How Do You Regulate a Self-Improving Algorithm?,” Atlantic, October 25, 2017, https://www.theatlantic.com/technology/archive/2017/10/algorithms-future-of-health-care/543825/.
However, the FDA has made it clear that it will not be bound by any of its own guidance documents: these only represent the FDA’s “current thinking.”56 FDA, “Guidances,” https://www.fda.gov/industry/fda-basics-industry/guidances In practice, however, guidance documents are “frequently cited in meetings with the stakeholders,” so that “over the last two decades, these documents have become ‘binding’ for all practical purposes.”57 “How Binding Are the FDA Guidance Documents? DOJ Says, Not Much,” FDAMap, March 22, 2018, https://www.fdamap.com/how-binding-are-the-fda-guidance-documents.html; citing Ronald M. Levin et al., “Federal Agency Guidance: Its Role in Agency Operations, Industry Compliance, and Litigation,” October 19, 2017, https://www.americanbar.org/content/dam/aba/events/administrative_law/2017/10/005_federal.authcheckdam.pdf.
The FDA’s pre-certification program has just begun, but uncertainty about pre-clearing firms and whether or how products are covered may weaken future generations of technological innovation. In addition, Congress may decide to grant the FDA more medical device user fees that may also have adverse consequences (as the fees will act as an additional barrier to entry) for potential market entrants. As described above, the loss of potential innovators decreases consumers’ health outcomes by limiting health-improving innovations.
Whether the FDA offers exemptions with enforcement discretion or no exemptions with many different enforcement categories, its rules will keep manufacturers guessing. The key issue is that any steps a manufacturer takes to improve a product, as well as any changes in the FDA’s “current thinking,” will cause uncertainty about which category the product belongs in and how the product will be regulated
Ultimately, firm certifications can lead to rent-seeking, whereby companies use political action to procure private benefits for themselves. 58 David R. Henderson, “Rent Seeking,” Library of Economics and Liberty, accessed February 8, 2019, https://www.econlib.org/library/Enc/RentSeeking.html. Firms that support the FDA (and, perhaps, are willing to hire former FDA employees) may increase the likelihood of their products being approved—and, correspondingly, of competitors’ products not being approved. Such circumstances would violate two of the conditions that allow nutrition innovations (and other innovations) to benefit us. Not only would they grant rents to rent-seekers, they would also shield the healthcare industry from disruption.
Potential Benefits of Health- and Nutrition-Related AI
Given that multiple lifestyle factors affect our health and that those factors interact with each other in many ways not yet understood, medical devices will likely be developed that incorporate more and more data from our daily lives. What will begin as nutrition advice will begin to incorporate more and more health, genetic and preferences that will effectively personalize our choices. The connections are illustrated in figure 1.
Figure 1. Personalized Health Devices
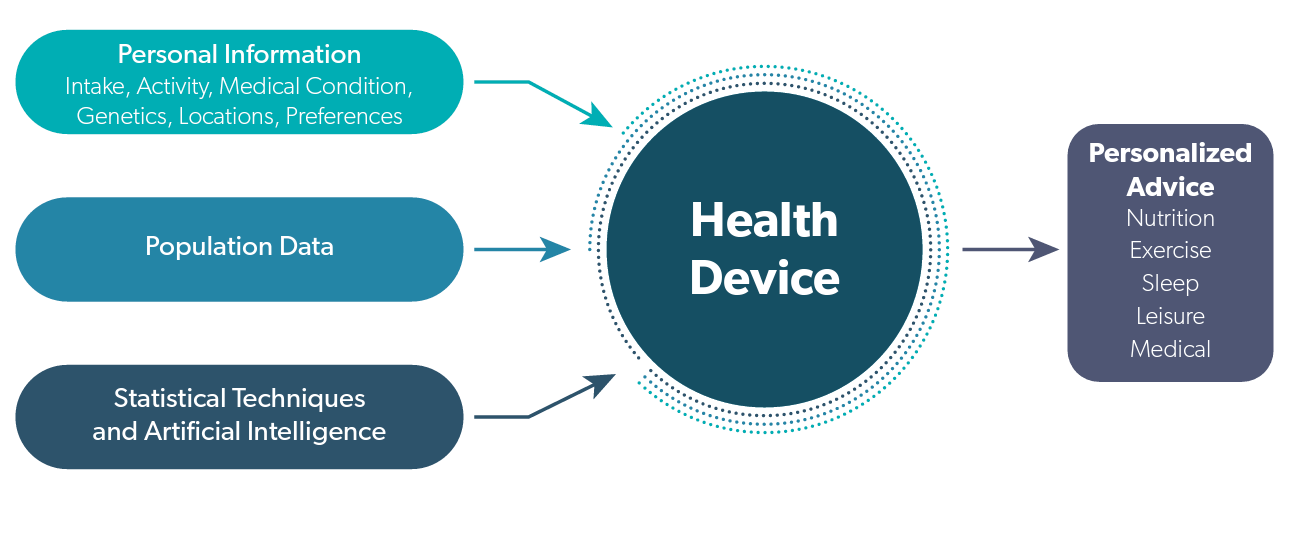
Personalized health devices will take all of the intakes, medical conditions, locations, activities and preferences for individuals and will combine that information with information on diet/disease relationships using population data and new statistical techniques to generate personalized advice.
However, regulations that penalize device manufacturers from using more information to make more useful health advice may result in adverse consequences.
For example, suppose that a person—let’s call him Jack—is predisposed to becoming diabetic and doesn’t know it yet. Jack uses a personalized health device that provides him with dietary recommendations. Clearly, Jack’s predisposition to diabetes should be a critical factor in his diet. In order to be most effective, Jack’s device should diagnose him as prediabetic. Unfortunately, if the device’s manufacturer advertises this capability, the device will no longer be exempt from the regulatory requirements for medical devices and the manufacturer may not be prepared to undergo the expenses and risks associated with this regulatory regime.
To keep the device from being regulated as a medical device, the manufacturer could program it to alter its dietary recommendations for people like Jack without actually giving them a diagnosis (or advertising such a capability). But in this case, Jack might not realize how serious his dietary choices are, and he might become diabetic. A diagnosis could also benefit Jack in other ways: for example, he might start using the drug Metformin,59 “Metformin for Prediabetes,” JAMA 317, no. 11 (2017): 1171 or he might change his exercise regime.NHS, “Pre-diabetes Prescription,” Prescription for Exercise, accessed June 21, 2018.
Suppose that Jack isn’t able to receive regular medical checkups, either because he can’t afford them or for some other reason. In this case, having a device that’s able to diagnose his predisposition to diabetes would be particularly useful. The cost of regulation weighs most heavily on low-income individuals who have the most to gain from innovations that make healthcare more affordable.
Personalized healthcare devices could also benefit consumers by helping people to combine food and drug intake to complement each other. Food and drugs interact, but—given that most physicians have little training in nutrition—a critical component of healthcare is missing. Nutrition consultant Mariëtte Abrahams describes how interactions of drugs with foods, particularly for those who take multiple drugs for their conditions, can change the composition of microbiota in the gut which can have long germ health implications.60 Mariëtte Abrahams, “The Personalised Nutrition Trend—How Digital Health Brands Can Revolutionise Healthcare,” MedTech Engine, May 16, 2017, https://medtechengine.com/article/personalised-nutrition/. With the help of new nutrition devices, interactions of drugs and nutrients can begin to be tracked. This sort of tracking goes beyond what most physicians do, yet it could be widely available to the general population.
Abrahams goes on to say,
- From bacteria in our poo, to metabolites in blood, or even the genes we have inherited from our parents, digital health brands need to ensure that we understand the person as a whole, as a system, in order to fine-tune nutrition and health advice. For this we need to track individual response to an intervention whether that comes from following specific dietary changes, to exercise, nutritional supplements or drugs for instance.61 Abrahams, “Personalised Nutrition Trend.”
The healthcare community has long recognized the interactive nature of external factors such as stress, socioeconomic condition, exercise, and diet and of internal factors such as genetic makeup and health status. Even so, the current regulatory regime in the United States disincentivizes manufacturers from creating devices that can track all relevant genetic, health and other significant information, diagnose diseases, and offer consumers advice to help them stay or get healthy. This is because creating such devices means jumping through regulatory hoops. In other words, there is a clash between what inventors want to invent—devices that have the potential to expedite our control over diseases—and the FDA’s approach of using fuzzy categories that impose ever more costly and time-consuming controls on innovators.
The FDA’s approach is in direct opposition to the creative approach of the entrepreneur. Medical doctor and health policy scholar Joseph Gulfo put it this way: “Due to ‘regulatory uncertainty,’ a euphemism for the complete and utter capriciousness and unpredictability in the FDA review process of new medical products, venture capitalists are becoming less inclined to fund very early stage companies.”62 Joseph V. Gulfo, Innovation Breakdown: How the FDA and Wall Street Cripple Medical Advances (Franklin, TN: Post Hill, 2014), pp 45-46 Yet, as explained below, more regulatory control that may also be accompanied by more uncertainty given the increased complexity is exactly what some authors are beginning to argue needs to happen.
Some Arguments for Greater FDA Control
Some commentators argue that more, not less, regulatory control of new devices is needed to keep consumers safe. Andrew Tutt, an attorney, argues that “algorithmic regulation will require federal uniformity, expert judgment, political independence, and pre-market review to prevent—without stifling innovation— the introduction of unacceptably dangerous algorithms into the market.”63 Andrew Tutt, “An FDA for Algorithms,” Administrative Law Review 69 (2017): 83–123
Tutt’s argument stems from an admirable and appropriate care for human well-being. However, his analysis fails to account for the reality that many of the technologies he describes—driverless cars, medical diagnostic imaging, distribution management—already have become not only cheaper than their human counterparts, but also more effective. 64 “Benefits of Imaging Using Radiation,” UCSF Department of Radiology and Biomedical Imaging, accessed February 9, 2019, https://radiology.ucsf.edu/patient-care/patient-safety/radiation-safety/benefits. Some of these technologies are now creating a safer society while reducing the cost of that safety. With such promising results for human well-being during the infancy of machine learning, to halt the progress of self-learning algorithms now could be as detrimental as it would have been to stop the specialization of labor when society was composed largely of subsistence farmers. This is not to suggest that there will be no problems with poor algorithms, but such problems are remediable.
It is important to understand the trade-off made when AI innovation is banned or stifled. The trade-off is not between the safety of the status quo and the possible downsides of a world in which AI’s potential has been realized. Instead, the trade-off is between the status quo—including both merits and perils—and the potential risks and rewards that AI and other innovations may bring.
Tutt points out that algorithms can fail “in ways we hardly could have predicted,” such as when IBM’s Watson answered the Final Jeopardy question incorrectly (naming Toronto as a U.S. city).65 Tutt, “FDA for Algorithms,” p.88. If we were comparing Watson’s improper answer to a world in which Final Jeopardy questions were answered correctly 100 percent of the time—or even 75 percent of the time—then Watson’s faux pas would be salient. Instead, the odds of a human being answering a Final Jeopardy question correctly are closer to 45 percent.66 The fan-curated J! Archive website calculated Season 33’s correct Final Jeopardy answers to be 306 out of 677 potential correct answers, or 45.2 percent. “Final Jeopardy! Round statistics for Season 33,” J! Archive accessed February 9, 2019, http://j-archive.com/finalstats.php?season=33. “Summary of Motor Vehicle Crashes,” Traffic Safety Facts: 2016 Data (DOT HS 812 580, National Highway Traffic Safety Administration, National Center for Statistics and Analysis, Washington, DC, September 2018). Focusing on Watson’s error—rather than on Watson’s overall performance in comparison to its human counterparts—misunderstands the trade-off in human versus machine skill.
Similarly, Tutt’s argument focuses on the downside of driverless cars but ignores their potential upside. The loss of any life is tragic, but fear of new technology’s risks should not prevent us from adopting innovations that will save 10 or 10,000 lives. The National Highway Traffic Safety Administration reported more than 7 million automobile crashes during 2016, over 34,000 of which were fatal.67 National Highway Traffic Safety Administration, “Summary of Motor Vehicle Crashes.” While more data are needed about the effects of different driving conditions, preliminary studies have found that driverless cars have lower crash rates and lower severe crash rates than traditional automobiles.68 Myra Blanco et al., “Automated Vehicle Crash Rate Comparison Using Naturalistic Data,” Virginia Tech Transportation Institute, January 2016, https://www.vtti.vt.edu/PDFs/Automated%20Vehicle%20Crash%20Rate%20Comparison%20Using%20Naturalistic%20Data_Final%20Report_20160107.pdf. In other words, even if driverless cars were to cause some otherwise-avoidable accidents, the damage would likely be vastly outweighed by the accidents they would prevent.
Attorney Matthew U. Scherer also argues for more regulation of AI. Like Tutt, he calls for an FDA-like governance structure that can pre-certify computer algorithms (particularly self-learning algorithms) before they are released into the market.69 Matthew U. Scherer, “Regulating Artificial Intelligence Systems: Risks, Challenges, Competencies, and Strategies,” Harvard Journal of Law and Technology 29, no. 2 (Spring 2016): 353–400. He also emphasizes unforeseen errors made by AI systems, stating, “a learning AI’s designers will not be able to foresee how it will act after it is sent out into the world.”70Scherer, 366. But that is the point of learning AI, what it learns is, by definition, unforeseeable.
Scherer calls for the courts to offer arbitration services, the legislature to offer guidance, and a regulatory agency to offer rulemaking and implementation of rules—essentially, for AI to be treated similarly to products now under FDA oversight. He states that “any AI regulatory regime must define what exactly it is that the regime regulates; in other words, it must define artificial intelligence. Unfortunately, there does not yet appear to be any widely accepted definition of artificial intelligence even among experts in the field.”71 Scherer, 359. If Scherer’s hypothetical AI regulator were to follow the precedent set by the FDA, it would likely expand its jurisdiction over time to protect against the possibility of harm from new AI products—but at the expense stifling innovation. Hence, consumers will suffer even greater harm by using existing inferior products.72 Numerous authors have pointed out that, because of the FDA’s authorizing legislation and its culture, it is often overly precautionary, preferring to make the mistake of preventing a good drug or device from coming to market rather than allowing a bad one from coming to the market.
Similarly, law professor Nathan Cortez writes, “Agencies need not be so deliberate and tentative with regulating innovations—even disruptive ones.” Instead, he suggests that agencies should be aggressive: he claims that most agencies underregulate and underenforce. In contrast to Tutt and Scherer, who call for the creation of a new regulatory structure for AI, Cortez notes that many of the devices fall under the purview of existing regulatory agencies.73 Nathan Cortez, “Regulating Disruptive Innovation,” Berkeley Technology Law Journal 29, no. 1 (Spring 2014): 175–228.
It would be easy for a legislative body (Congress) to cede its regulatory authority over algorithmic AI technologies to the administrative state (regulatory agencies). As Scherer notes, legislatures lack subject-matter knowledge, they often relinquish responsibility, and, when there are problems, they lay the blame for regulatory failures on the regulatory agencies.74 Scherer, “Regulating Artificial Intelligence Systems,” 379–80. However, given the natural inclinations of regulatory agencies to expand their jurisdictions, ceding power to regulatory bodies can seriously stifle innovation and, with it, positive effects on health, wealth, and well-being. Encouraging further regulation is not the best method for ensuring the safety of emerging devices.
A Positive Role for the FDA
At this point, it may seem that government has the capacity only to negatively influence the emerging market for nutrition devices. Government can have a positive role to play in this drama, however: the FDA can foster innovation by providing or validating biomarkers that will be needed to alert consumers when products are working to prevent or cure illnesses. Using validated biomarkers, consumers will be better equipped to judge on an individual basis whether devices are working to improve their personal health outcomes.
The issue with many biomarkers (and there are thousands of them) is that they have not been validated. Biomarkers should be public goods: once validated, they should be available to be used by anyone. The provision of biomarkers as public goods is an appropriate role for government, one in which government can protect consumers from bad science by ensuring that those who identify and test biomarkers use proper methodology and rigor.
In fact, in 2018 the FDA created a Biomarker Qualification Program to validate biomarkers for use in drug development75 US Food and Drug Administration (website), “Qualifying a Biomarker through the Biomarker Qualification Program,” updated August 2, 2018, https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/ucm535882.htm.—although arguably the FDA needs to enhance this activity by reallocating more resources away from other activities to speed it up.76 John Audette, “FDA-Approved Biomarker Panels: The Good, the Bad, and the Ugly,” Biomarker Trends (Amplion), February 26, 2015, https://www.amplion.com/biomarker-trends/biomarker-panels-the-good-the-bad-and-the-ugly/. Once validated, these biomarkers will hopefully be able to be used for AI devices related to nutrition or general health as well as for drugs. Biomarkers can give much quicker results than waiting to see if a long-term health outcome such as diabetes, cancer, and heart disease has been affected. This is a role in which the FDA can help ensure that medical devices are helping consumers achieve their desired health outcomes.
Conclusion: Governance of AI Devices
The huge increases in rates of obesity and diabetes are just one testament to our failure to use education and information to inform and influence our dietary choices. The effectiveness of new technologies to improve nutrition and health will ultimately depend on two things: (1) their ability to reduce the time cost of tracking dietary inputs and (2) their ability to aggregate complex information into easy-to-use behavioral recommendations. Older regulatory structures that continue to slow down market innovation for health devices must be re-evaluated.
The costs and uncertainties that come with a heavy-handed regulatory approach will erect barriers that slow progress by decades. For instance, policies that favor certain companies over others will create monopolies, limiting the favored companies’ incentives to innovate (and forgoing innovations from the disfavored companies). Regulations that create artificial and uncertain distinctions between categories of medical devices will “box in” inventors and manufacturers, limiting innovation and ultimately harming consumers.
If medical device innovators are left relatively unimpeded, progress will come in the form of more and more individualized products addressing individual wants and needs. Eventually, it will become impossible for the FDA (or any agency) to manage centralized preapproval of all these individualized products. Yet preapproval of new innovative products by risk class is exactly what some early commentators are requesting.
Nutrition is important to health, but it is only one piece of the puzzle. When we add in individualized data about genetics, environment, preferences, health conditions, and more factors, the puzzle becomes massively complex. That is where algorithms and large data sets will overcome human limitations. It is important not to continue to rely on inflexible barriers that challenge every technological advancement that offers the potential to improve health outcomes.
The FDA has the capacity to either help or hinder innovation in nutrition devices. It can help set the stage for these devices by assisting with the validation of biomarkers. Or it can harm consumers by blocking every marginal improvement in these devices. When the government regulates inputs, this does more than deprive consumers of real-time cost savings and improved product offerings: regulating inputs deprives current and future consumers of the gains from innovation that could have occurred had that innovation not been dampened. Technological advancements that could have improved consumers’ health and lowered their costs are simply absent. These types of losses are the most dangerous because they compound over time. However, improvements to regulatory systems bring benefits that can also compound over time. We return to our requirements for this to happen:
- Changes must be market-driven, not government-driven.
- The government must allow disruption in the healthcare industry.
- Those seeking rents should not be granted them.
- Governance must focus on actual harm as opposed to possible harm.
These requirements may seem radical given FDA’s long history. But these suggested changes to FDA’s governing laws and culture are likely to benefit consumers with a rapid increase in health technologies that is crucial to improving their health outcomes.